Answer of Dermatopathology Case 93
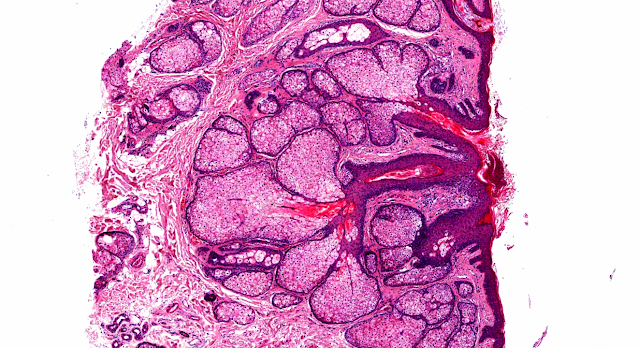
 Giemsa stain
Giemsa stain Toluidine Blue stain
Toluidine Blue stainVisit: Dermatopathology site
Image1 ; Image2 ; Image3 ; Image4 .
Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001 Jul;25(7):603-25.
The term 'mastocytosis' denotes a heterogeneous group of disorders characterized by abnormal growth and accumulation of mast cells (MC) in one or more organ systems. Over the last 20 years, there has been an evolution in accepted classification systems for this disease. In light of such developments and novel useful markers, it seems appropriate now to re-evaluate and update the classification of mastocytosis. Here, we propose criteria to delineate categories of mastocytosis together with an updated consensus classification system. In this proposal, the diagnosis cutaneous mastocytosis (CM) is based on typical clinical and histological skin lesions and absence of definitive signs (criteria) of systemic involvement. Most patients with CM are children and present with maculopapular cutaneous mastocytosis (=urticaria pigmentosa, UP). Other less frequent forms of CM are diffuse cutaneous mastocytosis (DCM) and mastocytoma of skin. Systemic mastocytosis (SM) is commonly seen in adults and defined by multifocal histological lesions in the bone marrow (affected almost invariably) or other extracutaneous organs (major criteria) together with cytological and biochemical signs (minor criteria) of systemic disease (SM-criteria). SM is further divided into the following categories: indolent systemic mastocytosis (ISM), SM with an associated clonal hematologic non-mast cell lineage disease (AHNMD), aggressive systemic mastocytosis (ASM), and mast cell leukemia (MCL). Patients with ISM usually have maculopapular skin lesions and a good prognosis. In the group with associated hematologic disease, the AHNMD should be classified according to FAB/WHO criteria. ASM is characterized by impaired organ-function due to infiltration of the bone marrow, liver, spleen, GI-tract, or skeletal system, by pathologic MC. MCL is a 'high-grade' leukemic disease defined by increased numbers of MC in bone marrow smears (>or=20%) and peripheral blood, absence of skin lesions, multiorgan failure, and a short survival. In typical cases, circulating MC amount to >or=10% of leukocytes (classical form of MCL). Mast cell sarcoma is a unifocal tumor that consists of atypical MC and shows a destructive growth without (primary) systemic involvement. This high-grade malignant MC disease has to be distinguished from a localized benign mastocytoma in either extracutaneous organs (=extracutaneous mastocytoma) or skin. Depending on the clinical course of mastocytosis and development of an AHNMD, patients can shift from one category of MC disease into another. In all categories, mediator-related symptoms may occur and may represent a serious clinical problem. All categories of mastocytosis should be distinctively separated from reactive MC hyperplasia, MC activation syndromes, and a more or less pronounced increase in MC in myelogenous malignancies other than mastocytosis. Criteria proposed in this article should be helpful in this regard.
Evolution of urticaria pigmentosa into indolent systemic mastocytosis: abnormal immunophenotype of mast cells without evidence of c-kit mutation ASP-816-VAL. Leuk Lymphoma. 2003 Feb;44(2):313-9.
Mastocytosis comprises a heterogeneous group of hematological disorders which are morphologically defined by proliferation and accumulation of tissue mast cells in one or more organs. Clinical manifestations of mastocytosis range from disseminated maculopapular skin lesions (= urticaria pigmentosa [UP]) that may spontaneously regress to highly aggressive neoplasms like mast cell leukemia or mast cell sarcoma. Recently, it could be shown that systemic mastocytosis (SM) is a clonal disorder often exhibiting mutations of c-kit, a protooncogene encoding the tyrosine kinase receptor for stem cell factor (SCF). Mutations of c-kit are considered to play a key role in the pathogenesis of mastocytosis. Therefore, we investigated the unique case of a 36 year-old male patient with indolent systemic mastocytosis (ISM) evolving from UP (cutaneous mastocytosis) by means of histology, immunophenotyping and molecular biology. At the time of initial diagnosis the bone marrow showed only a mild diffuse increase in mast cells but compact infiltrates were missing. The serum tryptase levels were normal. Five years later, however, the bone marrow histology displayed patchycompact mast cell infiltrates, which now allowed to establish the diagnosis of an ISM. The serum tryptase levels at this time were markedly elevated. At both time points, mast cells were analyzed by immunohistochemistry using anti-tryptase antibody AA1, by flow cytometry using antibodies against CD2 and CD25, and nested polymerase chain reaction (PCR) on laser-microdissected, single pooled mast cells. Immunohistochemistry revealed strong tryptase-positivity of mast cells in both cutaneous and bone marrow infiltrates. Flow cytometry yielded an aberrant expression of CD2 and CD25 on bone marrow mast cells. However, repeated thorough PCR analysis failed to unveil c-kit mutation in atypical mast cells of skin and bone marrow samples of both dates. These findings clearly show that ISM can evolve from UP. Moreover, our study provides further evidence that the c-kit mutation Asp-816-Val is not invariably present in ISM.
Mastocytosis: state of the art. Pathobiology. 2007;74(2):121-32.
Mastocytosis is a neoplastic disease involving mast cells (MC) and their CD34+ progenitors. Symptoms in mastocytosis are caused by biological mediators released from MC and/or the infiltration of neoplastic MC in various organs, the skin and the bone marrow being predominantly involved. A WHO consensus classification for mastocytosis exists, which is widely accepted and includes three major categories: (1) Cutaneous mastocytosis (CM), a benign disease in which MC infiltration is confined to the skin, is preferentially seen in young children and exhibits a marked tendency to regress spontaneously. (2) Systemic mastocytosis (SM) which is commonly diagnosed in adults and includes four major subtypes: (i) indolent SM (ISM, the most common form involving mainly skin and bone marrow); (ii) a unique subcategory termed SM with an associated non-mast cell clonal hematological disease (SM-AHNMD); (iii) aggressive SM usually presenting without skin lesions, and (iv) MC leukemia, probably representing the rarest variant of human leukemias. (3) The extremely rare localized extracutaneous MC neoplasms, either presenting as malignancy (MC sarcoma) or as benign tumor termed extracutaneous mastocytoma. Diagnostic criteria for mastocytosis are available and are widely accepted. SM criteria include one major criterion (multifocal compact tissue infiltration by MC) and four minor criteria: (1) prominent spindling of MC; (2) atypical immunophenotype of MC with coexpression of CD2 and/or CD25 (antigens which have not been found to be expressed on normal/reactive MC); (3) activating (somatic) point mutations of the c-kit proto-oncogene usually involving exon 17, with the imatinib-resistant type D816V being most frequent, and (4) persistently elevated serum tryptase level (>20 ng/ml). To establish the diagnosis of SM, at least one major and one minor criterion, or at least three minor criteria, have to be fulfilled. The natural clinical course of mastocytosis is variable. Most patients, in particular those with CM and ISM, remain in an indolent stage over many years or even decades, while others, in particular those with aggressive SM, SM-AHNMD, or mast cell leukemia, show a progressive course, usually with a fatal outcome.

Comments
Post a Comment